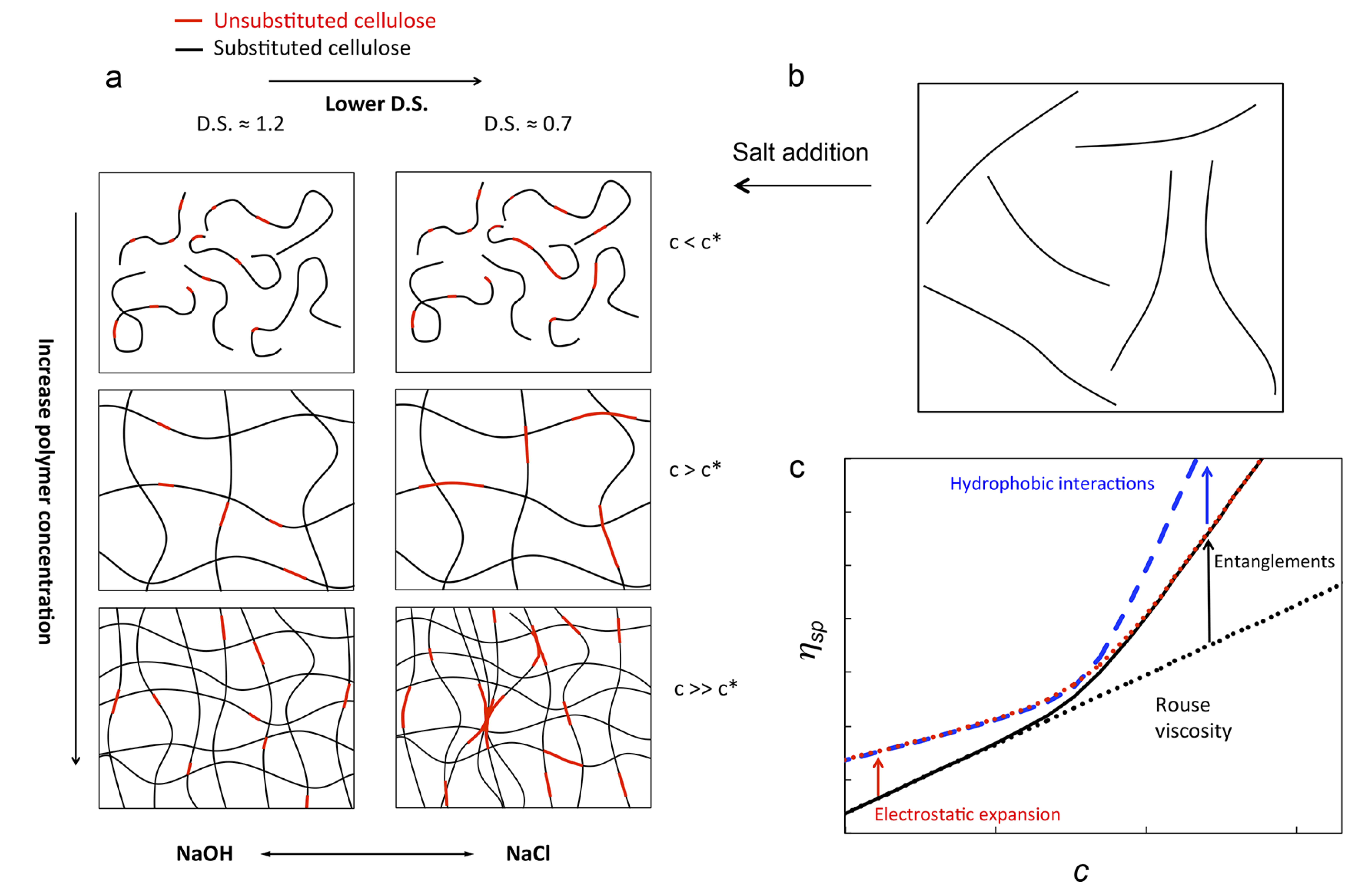
POLYMER SOLUTIONS AND GELS GROUP
Polyelectrolytes in solution
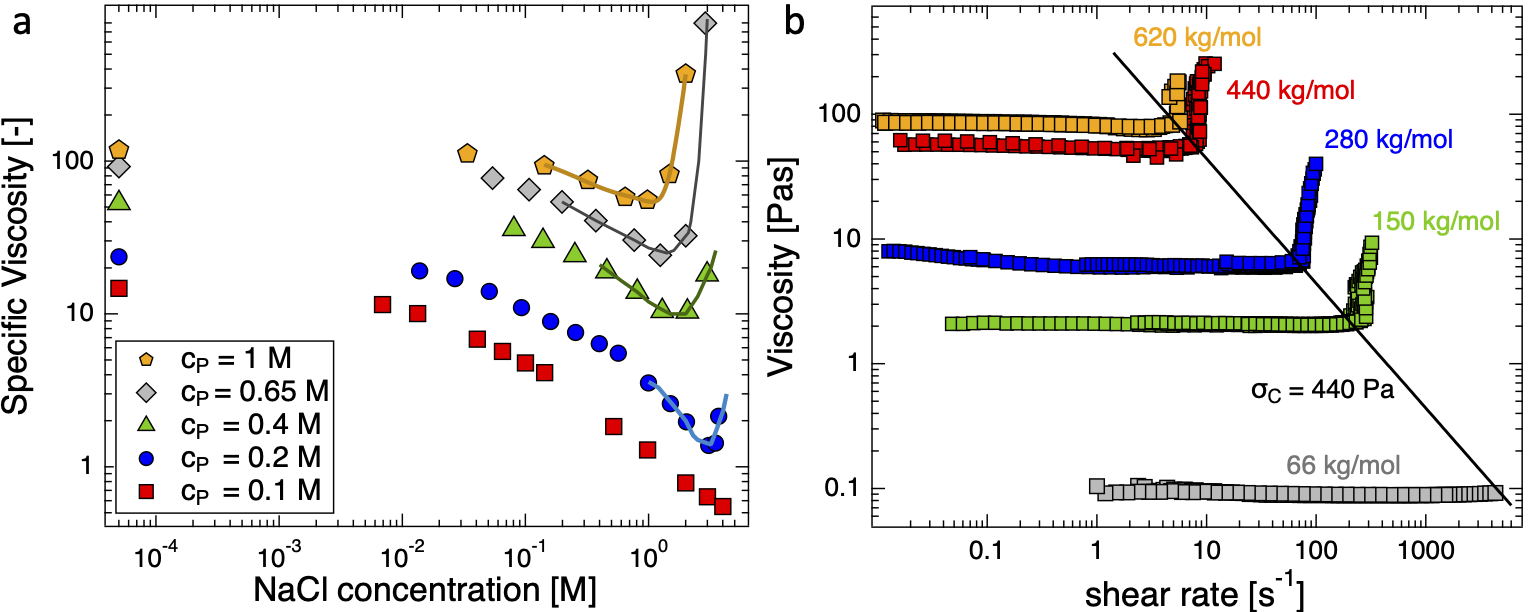
The central aim of our group's research is to understand the physics of polyelectrolyte conformation and dynamics in solution, a problem which we primarily tackle using scattering and rheological techniques. We apply scaling concepts to complex, multicomponent systems (e.g. polyelectrolytes in the presence of salts, surfactants or non-solvents) and extract structural and hydrodynamic properties.
Polyelectrolytes (PEs) are polymers with ionic groups along their backbone. In solution, counterions dissociate leaving the polymer with net charge, which makes them strongly correlated systems. Polyelectrolytes are present in the synovial fluid, where they provide lubrication between joints, in food products and pharmaceutical creams, where they act as texture modifiers. In wines, they can be added to prevent tartaric acid crystallisation, in laundry detergents they act as anti-soil redeposition agents and as water-softeners. Recently, mRRA (a polyelectrolyte) vaccines have transformed immunisation against COVID-19 on a global scale. Despite their enormous prominence in biological phenomena and industrial products, polyelectrolytes were once described by PG de Gennes as ‘the least understood form of condensed matter’, a designation which still holds true. Some of our research areas on polyelectrolyte solutions are outlined below. For overviews of some of the problems in polyelectrolyte physics, also check these reviews:
- Polyelectrolytes in dilute solution: Recent progress and open questions
- Rheology of Polyelectrolyte Solutions: Current Understanding and Perspectives
- Structure and linear viscoelasticity of flexible polymer solutions: comparison of polyelectrolyte and neutral polymer solutions
- Theory of polyelectrolytes in solutions and at surfaces
Counterion condensation
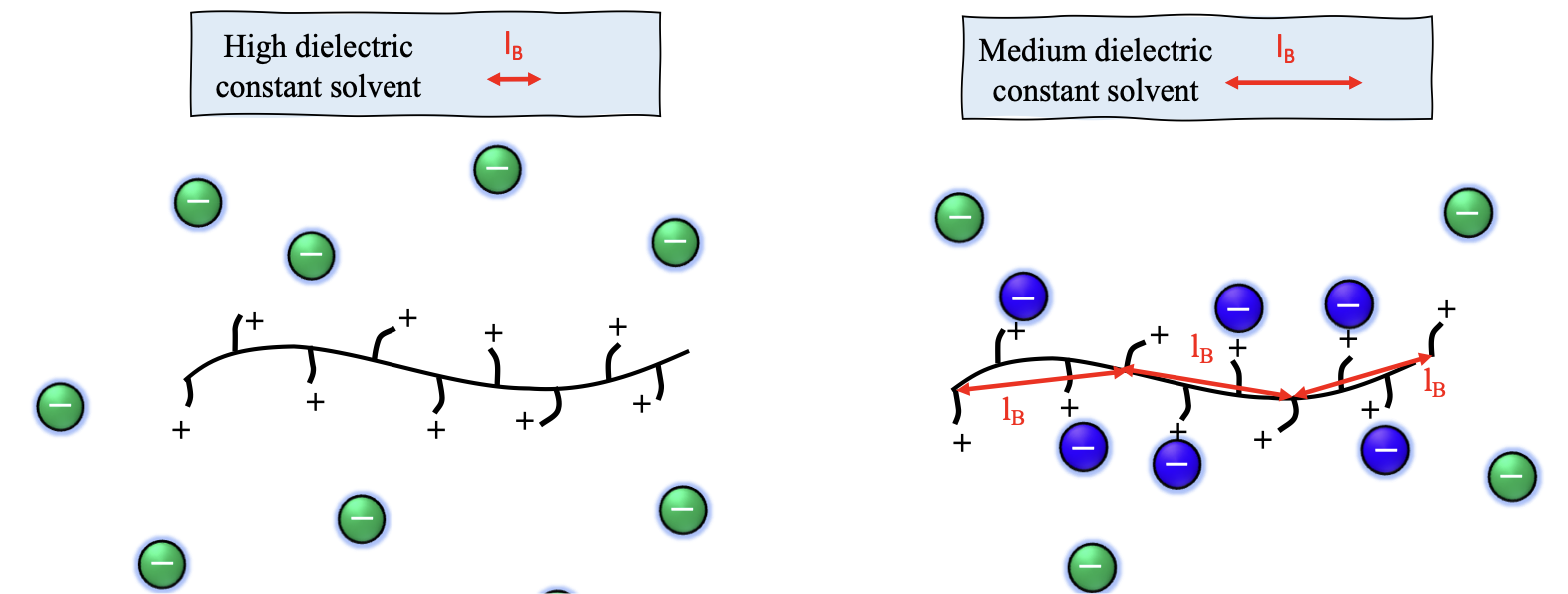
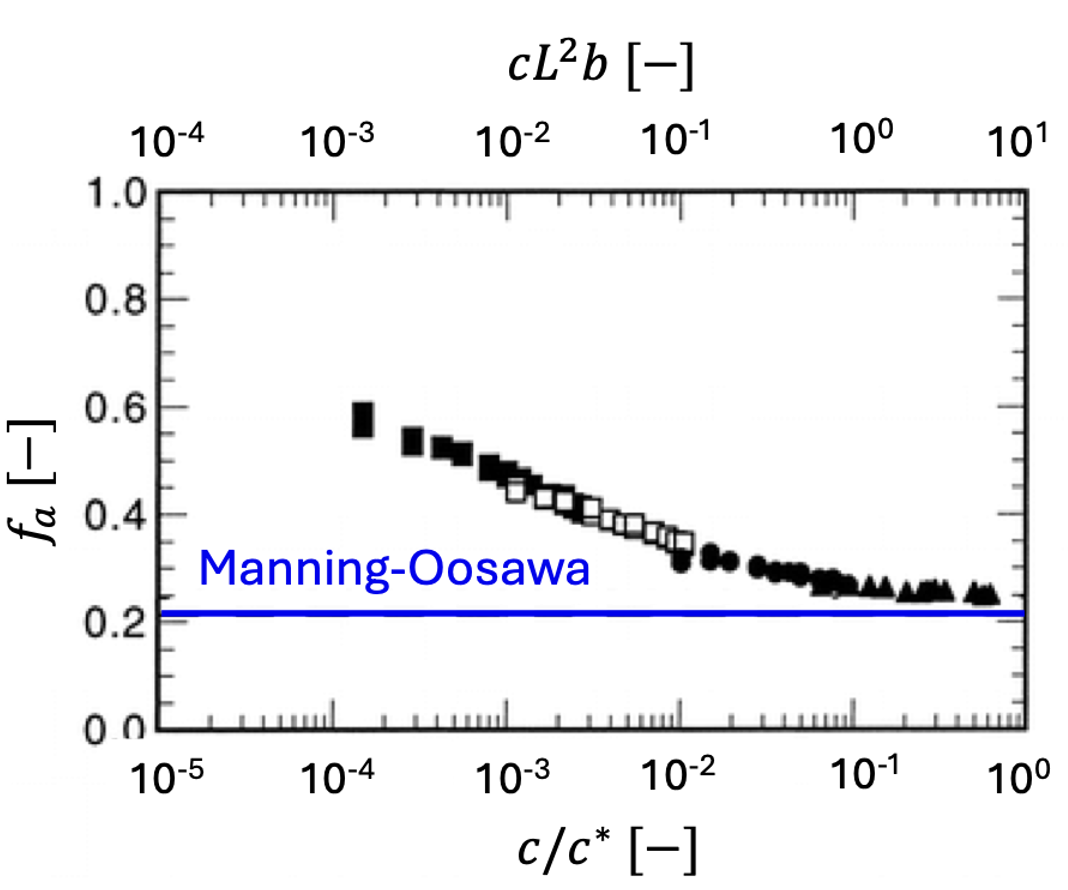
The Oosawa-Manning model was derived for rod-like chains at infinite dilution but experimental data plotted in figure 2 show that the OM threshold only applies above the overlap concentration c* (semidilute regime). Below c*, the counterion activity (\(f_a\)) in-creases on dilution. For \(c/c^*\ll 1\), it is expected to reach unity, as predicted by of Muthukumar and Dobrynin and co-workers. Tang and Rubinstein developed a model of counterion condensation which reproduces the Oosawa-Manning result above c^*
Within the framework of the Manning model, the fraction of condensed counterions (f) should be solely a function of:
\(\ \)
- the distance between charged groups
- the valence of the counterions
- the Bjerrum length of the solvent
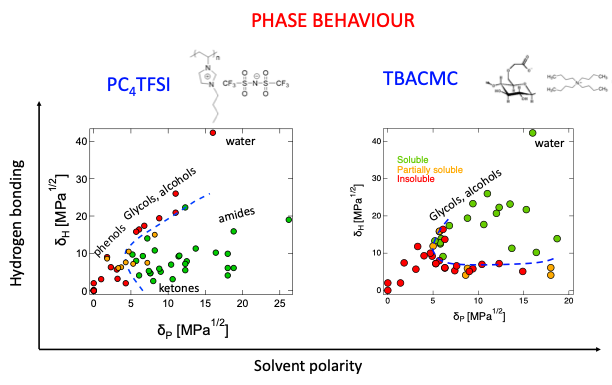
The nature of the counterion and its non-electrostatic interactions (e.g., hydrophobic effects) with the side group and backbone are not considered in the Oosawa–Manning model.
Counterion condensation vs. charge density: Several reliable datasets exist for the dependence of the fraction of condensed counterions (and related parameters) as a function of charge density. These have been reviewed by Manning in: [Manning 1979, Manning 1996, Manning & Ray 1998] and more recently by us [Gharehtapeh et al. 2025]. The predictions of Oosawa–Manning qualitatively agree with the experiments.
A major limitation of the current literature is that, despite the relatively broad set of systems that have been investigated, counterion condensation has not been measured for a single system with multiple techniques (e.g., electrophoresis, osmometry, conductivity). From the data available, it is clear that different methods yield different values for the fraction of condensed counterions. For example, osmotic, conductivity and dielectric spectroscopy estimates for the fraction of free counterions for NaPSS in water were compared in [Bordi et al. 2002], and disagree within a factor of ≈ 3. A comparison of six methods for estimating the effective charge density of carboxymethyl cellulose yielded similar disagreements [Gharehtapeh et al. 2025], presumably because different techniques probe different ion populations.
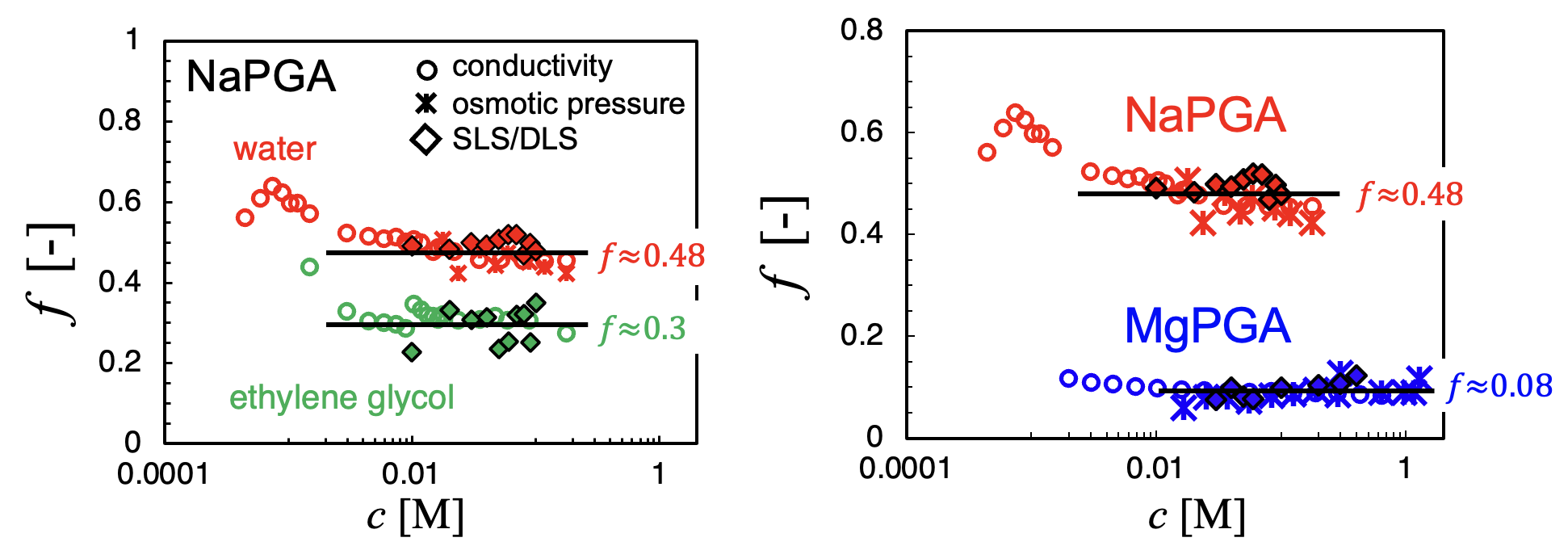
Influence of counterion valence: Potentiometric and osmometric measurements by Rinaudo and co-workers, reviewed by us in [Gharehtapeh et al. 2025], show that the effective charge fraction of polyelectrolytes with divalent counterions is roughly half of that with monovalent ions, in agreement with the Oosawa–Manning model. Data for trivalent counterions are still lacking due to the poor solubility of trivalent polyelectrolyte salts in water. In Section II, we present new results addressing this limitation.
Counterion condensation vs. Bjerrum length: Most studies on polyelectrolytes have focused on aqueous systems. Aqueous/organic mixtures have been studied, but only within narrow Bjerrum length ranges [Hou et al. 2025a]. The reason for this gap in experimental studies is likely the poor solubility of polyelectrolytes in organic media, see below. Direct measurements of the free-ion fraction (f) as a function of Bjerrum length, excluding such mixtures, are limited to three studies. Two of them yielded results inconsistent with Manning theory: [Lopez et al. 2024] inferred f from overlap-concentration scaling of two poly(ionic liquids), and [Beer et al. 1997] fitted a variational model to the radius of gyration data of quaternized poly(2-vinylpyridine) in several solvents. In contrast, the conductivity measurements of Gulati et al., 2025, which provide a more direct estimate of f, show better agreement with Oosawa–Manning predictions (see this preprint ).
In practice, obtaining reliable values of $f$ is not straightforward. Osmotic pressure measurements provide a direct estimate of the number of osmotically active ions but it is difficult to measure the osmotic pressure in organic solvents using commercial osmometers. Light scattering offers an attrative method to study the osmotic properties of polyelectrolyte solutions because the zero-angle scattering intensity is related to the osmotic compressibility of the solution. Specifically, the excess light scattering intensity (usually expressed as the Rayleigh ratio \(\Delta R\) is: \[ \Delta R(q) = K \rho_{\mathrm{pol}}^{2} N_A S(q) \] where \(\rho_{pol}\) is the polymer density, \(S(q)\) is the total structure factor and \(K\) is the optical contrast: \[ K = \frac{4\pi^2 n_0^2}{N_A \lambda^4}\left(\frac{dn}{dC}\right)^2 \] where \(\lambda\) us the wavlength of light and \(\frac{dn}{dC}\) the refractive index increment. The total structure factor at zero angle is: \[ S(0) = k_B T \, c \, \frac{dc}{d\Pi} \] and therefore allows, in principle to measure the derivative of the osmotic pressure with respect to concentration, from which the fraction of free counterions can be obtained (\(\Pi \simeq k_BTfc\)). In practice, however, applying this relation to salt-free polyelectrolytes is not straightforward because the scattering function at low $q$ is typically dominated by the so-called low-$q$ upturn. This excess scattering leads to values of the zero-angle intensity that are several orders of magnitude larger than those expected from the osmotic compressibility of the solution. The origin of this low$-q$ upturn is not understood and has been extensively debated in the literature. Recently, we showed that the low-$q$ upturn can be largely removed by filtration through sufficiently small pores and that the remaining contribution of the slow mode can be separated using dynamic light scattering (DLS), see the figure below. Once this contribution is removed, the resulting scattering intensity is found to correspond to the osmotic compressibility expected for salt-free polyelectrolyte solutions. The corrected zero-angle intensity can therefore be used to determine the fraction of free counterions.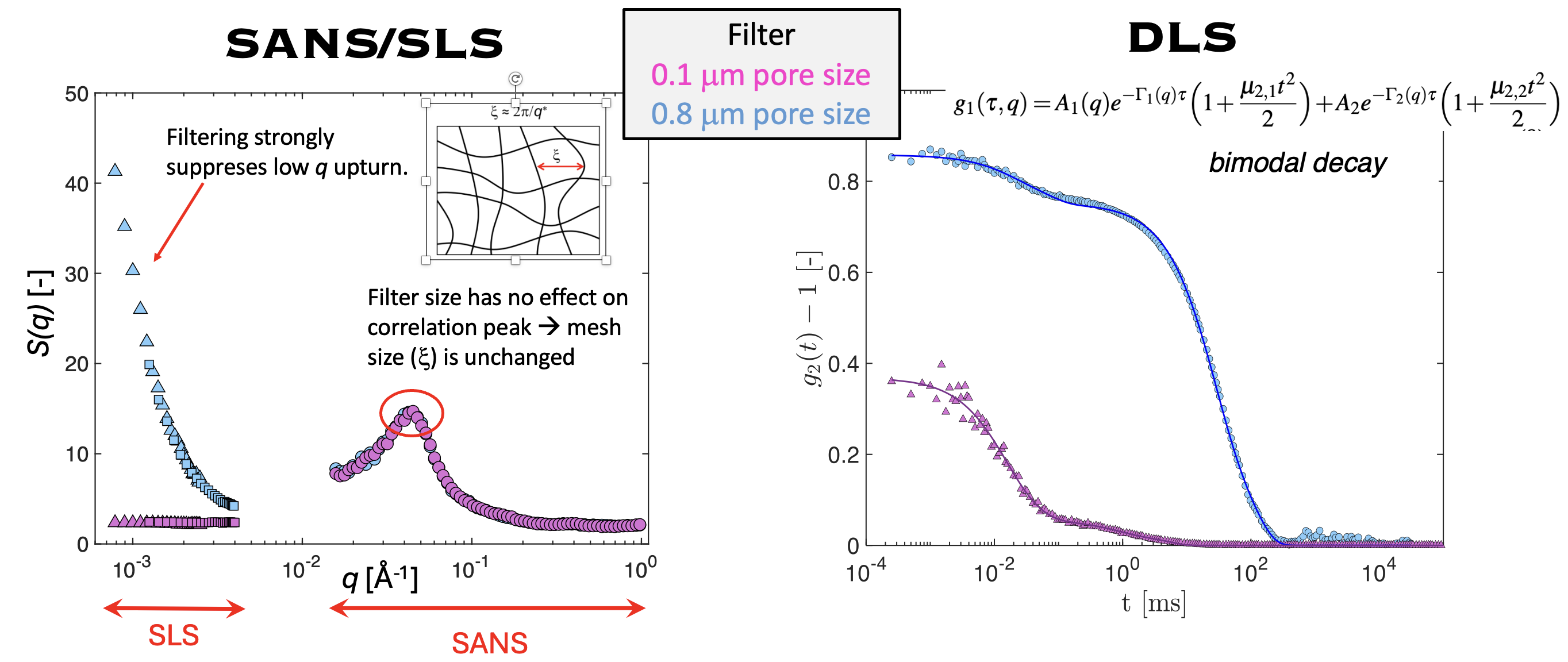
Ion pairing
The concentration of counterions within the condensed layer approaches the molar regime even if the overall ion concentration in the solution is low. At such high concentrations, ions can associate to form ion pairs. Unlike counterion condensation, which depends mainly on charge density, ion valence, and solvent dielectric constant, ion pairing is strongly influenced by the solvent structure around the ion. Ion pairing is therefore important for understanding specific ion effects, including ion selectivity, phase behaviour, and ion transport in polyelectrolyte solutions, gels, and membranes.
Our research quantifies ion pairing using density and speed of sound measurements. When ions associate with charged groups on the polymer, changes in hydration lead to measurable changes in the system's volume and the compresibility of the solvent. By analysing how density and sound velocity vary with ion type and concentration, we evaluate the extent of ion pairing and the corresponding hydration changes. We then use equilibrium dialysis to determine how strongly different ions are preferentially retained or excluded by the polymer. Combining these methods allows us to link microscopic ion pairing with macroscopic ion selectivity, providing a quantitative framework to understand and design selective polyelectrolyte materials.